
We have a definition problem in thyroid science and thyroid therapy.
A lot of confusion stems from the many definitions the words “euthyroidism,” “hypothyroidism” and “hyperthyroidism.”
These are terms that can be defined in many ways by doctors, patients, and even by thyroid researchers.
Is a person “hypothyroid” when their FT3 falls below their individual set-point? Or do they become hypothyroid only if their TSH rises above reference? Or is part of their body hypothyroid when their cardiovascular system decides to convert a lot more of its T4 into Reverse T3, and convert T3 into T2, causing a lack of T3 supply to nuclear receptors in heart muscle, resulting in heart failure?
The biggest problem today arises from viewing all through a narrow biochemical definition of thyroid status–usually dominated by TSH and Free T4 hormones and their reference ranges–while deemphasizing aspects of thyroid status that put biochemical features in context.

There are four ways of defining thyroid status:
- Etiology (cause)
- Gland health status
- Thyroid biochemistry
- Tissue thyroid hormone status
In this post, I illustrate that these four definitions are like “lenses,” ways of seeing aspects of thyroid status.
All of these definitions are necessary.
They’re like 4 lenses in a telescope.
Once we know we are looking at a thyroid hormone or thyroid gland disorder, all the lenses together can help us see the unique characteristics of an individual patient’s condition.
There cannot be a single definition because thyroid status is a complex phenomenon. A single patient may have
- more than one antibody that affects thyroid status,
- more than one type of thyroid / pituitary / hypothalamus gland disorder
- more than one thyroid biochemical deficiency or excess, and
- more than one tissue in their body that manifests thyroid status.
Even if a patient has a “simple” case of autoimmune hypothyroidism, these four aspects of thyroid status interrelate and help to explain what is going on.
Each definition or “classification” arises from different ways of measuring and diagnosing thyroid disease or thyroid imbalance. Each “lens” provides a different way of seeing thyroid status that helps us interpret what is going on in the body.
Many puzzling contradictions, symptoms and illnesses are left unresolved when we employ only a single-lens magnifying glass–our medical systems’ overreliance on TSH and FT4 to define thyroid biochemistry. This narrow lens has overwhelmed and silenced other pieces of information that are clinically relevant.
Using all four lenses offers hope for patients with many chronic diseases.
Combining these four definitions not only helps us treat thyroid disorders more effectively; it may also enable us to treat thyroid-hormone-influenced health disorders like cancers, heart diseases, and mental illnesses more effectively.
The origin of the definitions
These four definitions of thyroid status emerge from a broad and deep analysis of thyroid scientific literature from the 1950s to the present.
Seventeen years ago, in 2003, Fabrizio Monaco called for a “revision” in the “classification of thyroid diseases.” He noticed that despite the 1969 American Thyroid Association call to review classifications and terminology and update them as knowledge expanded, they had not done so for 30 years.
It appears that as of 2020, thyroid guidelines still teach doctors to define and treat thyroid diseases through outdated, narrow lenses.
Monaco proposed two major classifications, 1) Thyroid function, and 2) Clinical evolution. As he did so, he covered the four definitions covered in my article today.
- In the first class, “thyroid function,” he considered factors of
- etiology (genetics, autoimmunity) as well as
- gland health (thyroid goiter, pituitary failure).
- In the second class, “clinical evolution,” he considered
- biochemistry (“levels in the circulation”) as well as
- tissue thyroid metabolism and signalling “at the cellular level.”
However, by reading Monaco’s article today, one can see why his attempt at reclassifying failed. It was not flexible enough to permit non-hierarchical ways of seeing thyroid disorder characteristics, causes and effects.
By classifying gland disorders in a hierarchy, Monaco embedded contradictions in his model. How could “tissue hypothyroidism” include both diseases that resulted in hypothyroidism, conditions “without hypothyroidism,” and “transient hypothyroidism”? What??
Monaco was also left with two orphaned tissue manifestations he could not neatly fit into his taxonomy’s structure, namely “thyroid-associated ophthalmopathy” (Graves’ eye disease) and “Abnormal thyroid parameters without thyroid diseases (nonthyroidal illness, deficit of TBG, etc). Thyroid scientists seem to have the most discomfort with acknowledging that tissue hypothyroidism can coexist with thyroid gland health and a normal TSH.
Therefore, the order in which I list the definitions is not meant to be a hierarchy, but rather a matter of identifying what usually comes first, a physiological cause leading to an effect. It is not a chain reaction in which A must go through B to get to C and D. Rather, four definitions can overlap or bypass each other. Etiology A can also affect B, C, or D by independent pathways.
A more flexible four-lens definition model resolves Monaco’s orphaned classifications. For example, it is a model that can acknowledge that Graves’ disease antibodies may or may not manifest in elevated thyroid hormones, but they may nevertheless manifest in eye disease even when both thyroid hormones are in normal reference range and TSH is not low. Graves’ disease involves TSH-Receptor antibodies, and eye tissues have TSH receptors, and therefore “euthyroid Graves disease” exists and can nevertheless manifest in clinical disease (Watanabe et al, 1995). However, these patients may become hyperthyroid or hypothyroid next year or next month, depending on fluctuations in their antibody status (Takasu et al, 2012).
In addition, the final outcome of tissue hypothyroidism could in turn become an etiology/cause, enabling a circular model rather than a linear one. Kidney disease can act as an etiology of a low T3 state, because like any nonthyroidal illness, it may cause thyroid deiodinases in tissues to shift profoundly. But the fact that low T3 is an effect does not rule out the possibility that a chronic low T3 can cause or maintain both biochemical and tissue hypothyroidism that can, in turn, worsen kidney disease and subvert its therapies. It can be a vicious cycle.
I also chose this order to shift away from the point of view of the clinician and toward the point of view of the patient’s bodily experience over time. This is a perspective that Monaco gestured toward in his term “clinical evolution” but never quite succeeded, because it is an evolution only as seen and experienced by the clinician. The thyroid patient should not be viewed as a disconnected collection of organs, genetics, antibodies, and symptoms, and that’s often the way taxonomies cut us up into parts.
Even though a medical diagnosis hardly ever begins with etiology, the etiology is where every thyroid hormone problem begins. That is where we must go if we truly want to understand and address the cause and the glandular, biochemical, and tissue manifestations of a patient’s condition.
The fourth definition, tissue thyroid hormone status, is the most significant for human health. This is the true “clinical endpoint” of thyroid therapy which requires us to question the medical dominance of the “surrogate endpoint” of TSH normalization.
Tissue euthyroidism for the entire human body ought to be the goal of therapy and the judge of its success. The manipulation of antibodies, a gland’s health status, and biochemistry are means to achieve the true end of medicine.
#1. Etiology (cause)

Thyroid science offers descriptive adjectives that identify etiology distinguish one type of hypothyroidism or hyperthyroidism from another — for example,
- Postpartum thyroiditis versus Hashimoto’s thyroiditis.
- Graves hyperthyroidism versus Iatrogenic thyrotoxicosis.
- TSH-secreting Pituitary Adenoma causing thyrotoxicosis, versus hCG hormone-induced thyrotoxicosis during pregnancy.
An etiology is a cause that does not necessarily have an immediate effect, and it may never achieve its full effect.
For example, if you have high levels of thyroid peroxidase antibody (TPOAb), you may have thyroid autoimmunity, but you might not yet have lymphocytic infiltration (fibrosis) of the thyroid gland leading to slowly progressing gland hypofunction. Your gland may be responding well to higher levels TSH by secreting slightly more T3 to compensate for lower T4 production, and you might not yet require thyroid hormone replacement. (Hoermann et al, 2020)
Nevertheless, after an etiology clearly manifests itself in gland health, biochemistry, and/or tissue status, defining its etiology (or etiologies) can clarify and redefine the type of hypothyroidism you already have.
Etiologies involve factors such as:
- genetics that predispose one toward the production of anti-thyroid antibodies
- the level of one or more thyroid antibodies in circulation
- physical injuries to the pituitary and/or hypothalamus,
- genetic failures in hypothalamus pituitary TSH signalling
- medications and substances that can abnormally reduce TSH secretion from the pituitary
- severe genetic disorders in metabolism, transport or receptor sensitivity, which can cause problems even without damage or disease in the thyroid gland
- acute nonthyroidal illnesses and injuries like myocardial infarction, sepsis, heart surgery, which cause temporary biochemical hypothyroidism without a gland disorder
- chronic nonthyroidal illnesses like heart disease, depression, metabolic syndrome, which cause long term biochemical and worsen tissue hypothyroidism
- dietary choices like calorie restriction, fasting, or exhausting exercise that can skew TSH and T4-T3 conversion (Chatzitomaris et al, 2017)
Diagnosis of etiology is clinically useful information. It can predict the likelihood of dysfunctional responses to treatments, and even the desirable possibility of remission. Without this knowledge,
- Doctors may treat a temporary biochemical phenomenon as if it is something permanent, or vice versa.
- Doctors may treat only part of the patient’s disease without realizing that additional pathologies, such as pituitary failure or iodine overdose, are compromising the patient’s health.
Real life is complex. Etiologies overlap. A person may have Graves’ antibodies and Hashimoto’s antibodies in circulation at the same time, competing with each other or working in tandem. A person may have polymorphisms in DIO1 as well as a partial SPB2 deficiency and mild genetic pituitary resistance to thyroid hormone. These overlapping etiologies can make the clinical and biochemical presentation appear contradictory or confusing. This is why thyroid hormone biochemistry (definition #3) and gland status (definition #2) are not definitive enough.
Etiologies can derail therapies and cause illness. When a person with complex or undiagnosed etiologies is treated by merely normalizing their TSH when their TSH is not trustworthy, a person can be underdosed and become severely ill and disabled over time. The medical system currently casts the blame on the organs and tissues harmed by the failure. It is truly a failure in thyroid diagnosis and therapy.
#2. Gland health status

Thyroid status is so much more than just thyroid gland function.
Thyroid, pituitary, and hypothalamus gland health status have yielded the distinction between
- primary (thyroidal),
- secondary (pituitary) and
- tertiary (hypothalamic) thyroid disease.
The latter two are now categorized as “central hypothyroidism,” and they are among the most difficult and complex diagnoses in thyroid therapy today.
For example, as explained by Beck-Peccoz et al, 2017, tertiary hypothyroidism is the most difficult form of hypothyroidism to diagnose because “bioinactive” TSH molecules can’t stimulate a thyroid. Hypothalamic failure to secrete TRH or the inability of the pituitary to respond to TRH signalling causes an otherwise healthy pituitary gland to secrete inactive TSH molecules at a concentration that can even rise mildly above reference range. This can look just like the biochemistry of subclinical hypothyroidism.
However, if the thyroid gland is not responding normally to a TSH of 6.5 by enhancing its FT3:FT4 ratio appropriately, and if a thyroid ultrasound reveals a healthy normal thyroid, classifying the case as subclinical hypothyroidism is not helpful. The patient’s painful struggle with adrenal hypofunction and other disabling symptoms may be due to undiagnosed signalling and secretion problems in their hypothalamus and pituitary. Thyroid therapy may hinder if the nature of central gland dysfunction is not fully diagnosed.
The degree and type of gland disability influences a person’s degree of dependence on therapy–and the degree to which thyroid therapy can go terribly wrong if maladjusted.
The dysfunction of one or more glands can also significantly alter the overall thyroid hormone economy and the interpretation of test results during therapy.
Gland health status can be screened by TSH, FT4 and FT3 biochemistry if a person is not treated yet. For example,
- If TSH is extremely high or low, it can be extremely clear that a gland dysfunction is present, if a person is not dosing thyroid hormones. However,
- If TSH is at the borderlines of the reference ranges or within reference range, FT4 is necessary in addition to TSH to discern either pituitary or hypothalamic dysfunction.
- Measurement of FT3 can assess whether a failing thyroid is responding adequately to TSH stimulation by enhancing T3 synthesis and T4-T3 conversion, or whether Graves’ disease TSH-Receptor antibodies are either blocking or overstimulating TSH receptors, or non-bioactive TSH in central hypothyroidism is failing to enhance T3 secretion from the thyroid.
- In addition, FT3 can be used in context with TSH, FT4 and RT3 to detect an undiagnosed chronic nonthyroidal illness in non-hospitalized patients in patients with or without a healthy thyroid gland.
However, once a person is on thyroid therapy, it is often difficult to discern gland health from biochemical patterns because of their mutual influence.
Science is finally coming to acknowledge that the degree of one’s gland health affects one’s response to standard thyroid therapy.
- Thyroidless patients are, in fact, biochemically different from other people because of their gland loss. Those without thyroid glands usually have poorer T4-T3 conversion within a normalized TSH (Midgley et al, 2015).
- Patients with autoimmune thyroid gland failure are also different from patients with pituitary failure who have healthy thyroid glands, because the healthy thyroid gland can convert T4-T3 more efficiently. (Sesmilo et al, 2011)
Ultrasounds provide important confirmations of gland health far beyond the diagnosis of nodules, goiter, and thyroid cancer grading. They determine the degree to which T3 hormone levels are vulnerable.
One of the most revolutionary findings in recent thyroid science is that thyroid gland health significantly influences T4-T3 conversion, not just T4-T3 synthesis/secretion.
Without a healthy thyroid gland “shielding” our FT3 levels through variable secretion and conversion, our FT3 levels will vary in relationship to TSH and FT4 in ways that we never see in nature.
As shown in the graph by Hoermann et al, 2016a, the range of FT3 levels in thyroid therapy drops significantly lower than the norm when TSH is at 0.0 standard deviations of the norm (the healthy population average). The loss of a thyroid gland and initiation of LT4 monotherapy requires TSH to be 2.5 standard deviations below the norm to achieve an equal FT3 with the healthy-thyroid population.

Loss of a thyroid gland turns the T3:TSH biochemistry ratio on its head in such patients, as shown in the same graph. The relationship between TSH and FT3, which is mildly a positive relationship in thyroid gland health, becomes a powerfully inverse relationship after the thyroid gland is removed and “replaced” only with Levothyroxine.
Therefore, the thyroid gland is not just a secretor of thyroid hormone at a static ratio of 14 parts T4 to 1 part T3, as some suppose.
Even Pilo et al’s 1990 article showed a wide range of diversity in thyroid secretion ratios among the 14 iodine-overdosed adults in their study. The thyroidal secretion rate (SR) of T3 and of T4 was as low as 1:6 in one patient, and as high as 1:71 in another. These individual differences compensated for lower to higher rates of T4-T3 peripheral conversion.
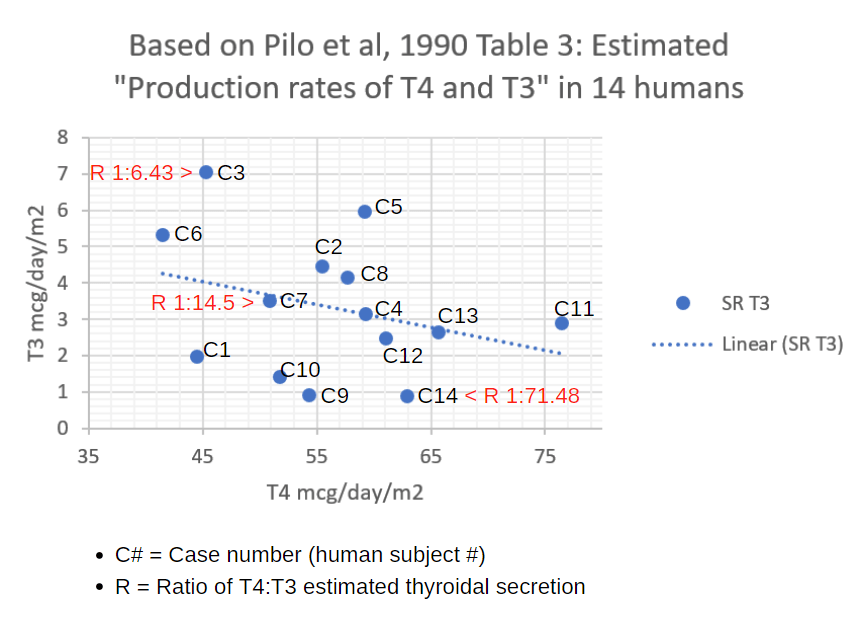
The thyroid gland is a flexible engine of thyroid gland synthesis AND conversion, and its overall secretion rate (SR) and T3:T4 ratio of secretion adjusts powerfully in response to TSH, iodine status, and its own health status.
In 2014, Abdalla & Bianco trumpeted studies showing that in mice, a healthy thyroid gland protects them from suffering from a complete failure in the genes responsible for T4-T3 conversion throughout the body.
The thyroid gland’s variable secretion ratio is a shield against underlying thyroid metabolic disabilities, since “the thyroidal content of T3 but not T4 is markedly increased … a factor that likely contributes to the maintenance of the serum T3 level” (Galton et al, 2009).
The thyroid gland doesn’t just take orders from TSH, but it protects the body from genetic defects in thyroid hormone metabolism by continually readjusting our T4:T3 ratio.
In hyperthyroid cases, additional diagnostic tests like iodine uptake scans and ultrasounds help to determine gland health. Gland health diagnostics are necessary to discern the source(s) of thyroidal overproduction:
- TSH-Receptor stimulating antibody
- Hyper-secreting thyroid nodules
- Rare thyroid gland infections (Subacute thyroiditis)
- Hashitoxicosis can cause temporary hyperthyroidism (Shabaz et al, 2018).
In cases where the thyroid gland is already damaged by Hashimoto’s or partial thyroidectomy, Graves’ stimulating antibody activity might not result in elevated thyroid hormones, but can still significantly alter biochemistry and cause TSH, FT4, and FT3 fluctuations that can derail hypothyroid therapy.
#3. Thyroid biochemistry

Biochemistry is the most common way of defining thyroid status. However, within this domain, heated debate and definitional diversity exists. Research scientists and/or guidelines have often arbitrarily chosen either a single hormone or two of hormones to define a biochemically hypothyroid or thyrotoxic state.
- TSH alone (often used in large population studies and in screening, but it is incapable of diagnosing central hypothyroidism, NTIS, or detecting ineffective thyroid therapy)
- TSH + FT4 together (the basis of definitions in American Thyroid Association guidelines since the 1990s, which do not discern sufficiently between screening prior to therapy and monitoring effective therapy)
- TSH + FT4 + FT3 together (in more advanced studies of thyroid therapy and relationships between thyroid hormones and various diseases)
- TSH + FT4 + FT3 + RT3 (the latter is usually only measured in studies of nonthyroidal illness. In clinical practice, it is employed by doctors to troubleshoot and optimize thyroid therapy when it includes T4 hormone)
- FT3 alone (This is often the sole relevant hormone test in T3 monotherapy, when FT4 and TSH may be normal, low or entirely suppressed by T3 dosing effects, while FT3 may be high-normal or elevated.)
- T4 / FT4 alone (historically, T4 / FT4 was prominent in thyroid science before TSH tests became more sensitive and rose in priority; Protein Bound Iodine, PBI was an older way of estimating T4 in blood)
Therefore, even within the definition of “biochemistry,” are up to six ways to define a person as “hypothyroid” or “hyperthyroid.”
Biochemistry is also plagued by the dominance of reference range boundaries. It is a plague because a ridiculously small difference of 0.1 pmol/L can misclassify a single patient as “euthyroid” if a local laboratory’s population statistics did not properly screen blood samples and remove those which had antibodies or which were from patients with chronic nonthyroidal illness. Researchers too often divide patient groups into cohorts by reference boundaries, wrongly assuming that they are clinically significant just because they create patterns of statistical significance.
Biochemistry need not be plagued by reference boundaries. Any two or more hormone levels may also be assessed in terms of their ratios, changes over time, and relative levels in relation to control groups. The laboratory reference boundaries can then be merely background information rather than arbitrary boundaries imposed on the analysis and interpretation of data.
A fundamental paradigm shift in thinking about thyroid biochemistry was clearly articulated by Abdalla and Bianco in 2014 when they declared that the main goal of thyroid hormone economy is not to normalize TSH but to “defend plasma T3.”

There is no single T3 level in bloodstream that is optimal for all humans, just as there is no single TSH that is optimal. The T3 level must be adjusted to the level that will obtain optimal health in the individual organism under the current state of metabolic stress they are facing.
When FT4 falls low in iodine deficiency or in early thyroid gland failure, FT3 must rise to compensate.
And what enables this defense? The healthy thyroid gland in cooperation with a healthy pituitary and hypothalamus.
What can we do with this mess of thyroid biochemical definition disagreement?
- First, stop isolating the TSH and FT4 from the FT3. These hormones describe a shifting triangle within an individual, not three separate hormones on a bar graph. (Hoermann et al, 2016b)
- Secondly, stop using TSH and FT4 to excuse or trump a low FT3. The Free T3 hormone level can often be the most powerful factor in tissue-level thyroid hormone signaling (Bianco et al, 2019).
- Thirdly, stop imagining that either TSH or FT4 can predict or ensure the optimization of FT3 within thyroid therapy, since therapies directly manipulate and distort these hormone relationships. The logic of monitoring thyroid therapy should not be the same as screening for thyroid disease in the untreated person (Hoermann et al, 2016b).
- Finally, stop isolating biochemistry from the three other definitions of thyroid status, and view the patient through all lenses together.
For example, the biochemical pattern of “nonthyroidal illness syndrome” (NTIS) or Low T3 Syndrome has been mislabeled as “Euthyroid sick syndrome” because the TSH in normal range makes the patient appear to be euthyroid. But scientists like Van den Berghe have been trying for decades to encourage us to differentiate “Acute” nonthyroidal illness from “chronic” nonthyroidal illness. Finally, people are questioning why some people fail to recover from nonthyroidal illness, and they are suspecting it is due to the failure to elevate TSH, a gland failure in the hypothalamus. They are also understanding how chronic NTIS can shift local tissue thyroid hormone status (Van den Berghe et al, 2014).
If one wishes to save patients from dying after expensive heart surgeries and chemotherapies for cancer, one must understand how biochemistry affects, and is affected by, tissue-level thyroid hormone metabolism
#4. Tissue thyroid hormone status

The ultimate definition of thyroid health is tissue euthyroidism.
Increasingly, thyroid science is coming to acknowledge that each tissue and organ in the human body metabolizes T4 and T3 thyroid hormones in different ways and at different rates.
Therefore, if you want to treat heart disease or cancer, you have to understand how thyroid hormones operate uniquely on heart cells and cancer cells. You have to understand how thyroid hormone metabolism can distort local thyroid hormone levels. Cancer tissues highly express Deiodinase Type 3 (Davis et al, 2018), causing localized low T3 in cancer tissue. Meanwhile, other tissues and organs may have sufficient T3 levels.
Even Reverse T3, long thought to be an inactive hormone, is now understood to be capable of non-genomic signaling in a receptor on the cancer cell membrane, alongside T4 hormone, to promote cancer proliferation (Lin et al, 2019).
Throughout the body, thyroid hormone T3 signaling in the nucleus of cells does most of the work, and secondarily, non-genomic T3 signaling in mitochondria and T3 and T4 signaling at cell membrane receptors, control the body’s local and global manifestations of thyroid hormone deficiency, excess or balance.
However, saying that each tissue differs in its thyroid hormone metabolism does not mean that bloodstream levels of thyroid hormone are irrelevant to human health or as guides to thyroid therapy.
A major review article by Bianco et al in 2019 amplified the reciprocal importance of bloodstream T3 and T4 on the one hand, and tissue thyroid status on the other hand. They are two sides of a coin, inseparable.
Bloodstream ratios and levels of FT3 and FT4 can profoundly influence, and are influenced by, derangements in tissue-level thyroid hormone conversion and receptor signalling.
Thyroid hormone transport is two-way. Free T3 in bloodstream does not just provide hormone supply to cells, but the Free T3 in blood is partly a product of intracellular hormone metabolism.
FT3 and FT4 (and RT3) concentrations in blood simultaneously reveal both supply from the thyroid (or thyroid hormone dosing) and the net effect of global tissue thyroid hormone metabolism.
In tissues, both sides of the “waterfall” of thyroid hormone conversion matter.
- On the one side of the cascade is the conversion of T4 to Reverse T3, and T3 to 3’3 T2.
- On the other side is the conversion from T4 to T3, and T3 to 3,5-T2.
When one side of the cascade dominates over the other at the wrong time, tissue thyroid hormone imbalance and pathology is the result.
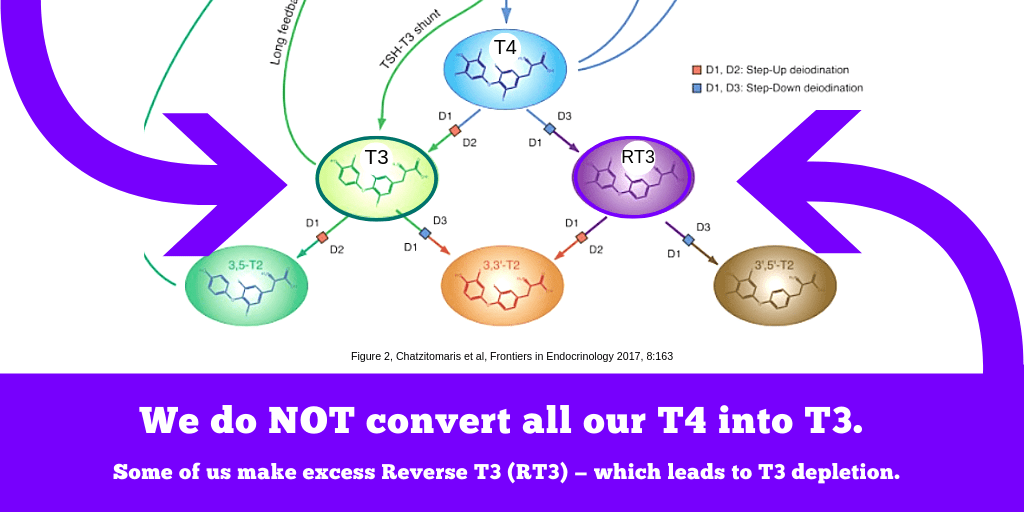
Beyond T3 and T4 levels, we can also “interview” each organ or tissue individually to discern how thyroid hormone signalling is having effects there, as long as you also understand the other factors that combine with thyroid hormones to modulate these biochemical signals. Prominent tissue-level thyroid hormone response biomarkers and signs include:
- pituitary TSH,
- liver ALT
- total cholesterol,
- kidney GFR,
- creatine kinase
- heart rate,
- body temperature
- ankle reflex
- mental health assessment instruments
- symptom scores
All of these tissue responses can be increased or decreased significantly by changes in T3 signaling in local tissues.
These and many other signs have been used in many studies that seek to understand tissue thyroid hormone signalling beyond the pituitary response of TSH secretion. (Celi et al, 2010, 2011; Escobar-Morreale, 2005; Meier et al, 2003; and Yavuz et al, 2013)
Tissue thyroid status is a way of defining euthyroidism that puts into question the overemphasis on biochemical reference range boundaries defined by a thyroid-healthy population.
In particular, studies in T3 monotherapy, T4 monotherapy, and nonthyroidal illnesses have revealed the inability of TSH and reference ranges to define tissue euthyroidism.
- Both within and outside of thyroid therapy, hypothyroid tissue status can coexist with a normalized TSH (Ito et al, 2012, 2017, 2019).
- A wide range of health conditions can induce low TSH without tissue thyrotoxicosis or prevent the elevation of TSH as a signal of tissue hypothyroidism (Chatzitomaris et al, 2017).
- The TSH reference range fails to be protective of tissue euthyroid hormone status in all thyroid patients (Ito et al, 2017; Larisch et al, 2018).
Tissue euthyroidism in LT4 thyroid therapy is associated with mid-range or higher T3 hormone levels in therapy and lower levels of TSH. In LT4 monotherapy a higher-than-reference FT4 and lower-than-reference TSH to achieve FT3 levels high enough to obtain true tissue euthyroidism (Ito et al, 2017; Larisch et al, 2018).
In addition, customization of individuals’ FT3 levels to remove symptoms of hypothyroidism and hyperthyroidism is possible in LT4 monotherapy (Hoermann et al, 2019), showing that in thyroid therapy, even small shifts in FT3 levels can influence tissue manifestations of thyroid disease.
Tissue response to T3 thyroid hormone alone can be most strongly discerned in humans who concurrently lack all T4 hormone. Celi, Yavuz and colleagues conducted a double-blind controlled study comparing LT4 monotherapy with LT3 monotherapy in thyroidless individuals. They wrote three very detailed and deep articles about the experiment (Celi et al, 2010, 2011, and Yavuz et al, 2013). At the end of their third publication, they insightfully remarked that TSH alone cannot define global tissue euthyroidism in either form of therapy:
- “the data suggest that pituitary euthyroidism, both assessed by basal or TRH-stimulated TSH, does not necessarily equate to a state of generalized euthyroidism at the level of the different targets of the hormonal action.” (Yavuz et al, 2013)
Low TSH is not synonymous with tissue thyrotoxicosis, either.
Researchers who understand genetic problems with thyroid deiodinases realize that in the most severe case, the “partial SBP2 deficiency,” patients require T3 hormone therapy. Their dose of T3 may not normalize FT4 or TSH, but may result in benefits to bone metabolism in a developing 10-year-old individual (Dumitrescu et al, 2010).
Even Triac therapy (Triac is an acetic acid derivative of T3 hormone) can suppress TSH benignly without inducing thyrotoxicosis, and it is therefore used therapeutically in patients who have genetic receptor insensitivity to T3 thyroid hormone (Groeneweg et al, 2017).
The T3 hormone is powerful as well as fragile at the tissue level.
Our T3 supply in blood can be depleted locally by derangements in metabolism and signaling–at any level of TSH.

Tissue euthyroidism, the red circle in the model, ought to be the ultimate goal of thyroid therapies.
Tissue euthyroidism is synonymous with health outcomes.
If we see the connectivity of thyroid status from 1) etiology to 2) gland health, to 3) biochemistry, to 4) tissue thyroid status, it’s clear we can’t stop at biochemistry and say “your TSH is normalized — you’re euthyroid.”
The label “euthyroid,” when it rests on such a thin foundation, closes doors and closes minds before questions can be asked. It sends a message that we’re all fixed in the thyroid department. Now attention turns to our diabetes or depression.
This is a horrible mistake. It ruins lives and costs our health care system and society in the long run.
Even if we do have a joint disease, a skin disease, or a mental health problem in addition to a thyroid disorder, the severity of these “tissue-based” disorders can be reduced, and our medications for them can be enhanced, by removing tissue thyroid hormone imbalance. When “tissues” are euthyroid, we can be more easily set free from symptoms and signs of hypothyroidism and hyperthyroidism in our joints, our digestion, our brain, our reproductive system, and our skin.
Here’s where thyroid patients need the most help from compassionate doctors: Putting TSH back in its proper place as only one of many possible ways of seeing / defining thyroid status.
- Listen to the paradigm shift in thyroid science and look beyond mere TSH normalization to define our thyroid hormone status.
- Realize that in many forms of thyroid disease and therapy, TSH concentrations can neither control nor predict T3 sufficiency in bloodstream or other tissues in the human body.
- Enable and honor individual thyroid patients’ achievement of tissue euthyroidism even when it induces “excursions” outside the TSH, FT4 and FT3 reference ranges. When many aspects of a person’s thyroid status are abnormal, an abnormal biochemistry can be therapeutic if it effectively compensates for those abnormalities.
Leave a public reply here, on our website.